Including inter-species measurements in differential expression analysis of RNAseq data with the compcodeR package
Paul Bastide & Mélina Gallopin
2026-04-28
Source:vignettes/phylocompcodeR.Rmd
phylocompcodeR.Rmd
library(compcodeR)
#> Loading required package: sm
#> Package 'sm', version 2.2-6.0: type help(sm) for summary informationIntroduction
The compcodeR R package can generate RNAseq counts data and compare the relative performances of various popular differential analysis detection tools (Soneson and Delorenzi (2013)).
Using the same framework, this document shows how to generate “orthologous gene” (OG) expression for different species, taking into account their varying lengths, and their phylogenetic relationships, as encoded by an evolutionary tree.
This vignette provides a tutorial on how to use the “phylogenetic”
functionalities of compcodeR.
It assumes that the reader is already familiar with the compcodeR package vignette.
The phyloCompData class
The phyloCompData class extends the
compData class of the compcodeR
package to account for phylogeny and length information needed in the
representation of OG expression data.
A phyloCompData object contains all the slots of a compData
object, with an added slot containing a phylogenetic tree with ape
format phylo, and a length matrix. It can also contain some
added variable information, such as species names. More detailed
information about the phyloCompData class are available in
the section on the phylo data
object. After conducting a differential expression analysis, the
phyloCompData object has the same added information than
the compData object (see the result object in the
compcodeR
package vignette).
A sample workflow
The workflow for working with the inter-species extension is very similar to the already existing workflow of the compcodeR package. In this section, we recall this workflow, stressing out the added functionalities.
Phylogenetic Tree
The simulations are performed following the description by Bastide et al. (2022).
We use here the phylogenetic tree issued from Stern et al. (2017), normalized to unit height, that has species with up to 3 replicates, for a total number of sample equal to (see Figure below).
library(ape)
tree <- system.file("extdata", "Stern2018.tree", package = "compcodeR")
tree <- read.tree(tree)Note that any other tree could be used, for instance randomly
generated using a birth-death process, see e.g. function
rphylo in the ape
package.
Condition Design
To conduct a differential analysis, each species must be attributed a condition. Because of the phylogenetic structure, the condition design does matter, and have a strong influence on the data produced. Here, we assume that the conditions are mapped on the tree in a balanced way (“alt” design), which is the “best case scenario”.
# link each sample to a species
id_species <- factor(sub("_.*", "", tree$tip.label))
names(id_species) <- tree$tip.label
# Assign a condition to each species
species_names <- unique(id_species)
species_names[c(length(species_names)-1, length(species_names))] <- species_names[c(length(species_names), length(species_names)-1)]
cond_species <- rep(c(1, 2), length(species_names) / 2)
names(cond_species) <- species_names
# map them on the tree
id_cond <- id_species
id_cond <- cond_species[as.vector(id_cond)]
id_cond <- as.factor(id_cond)
names(id_cond) <- tree$tip.labelWe can plot the assigned conditions on the tree to visualize them.
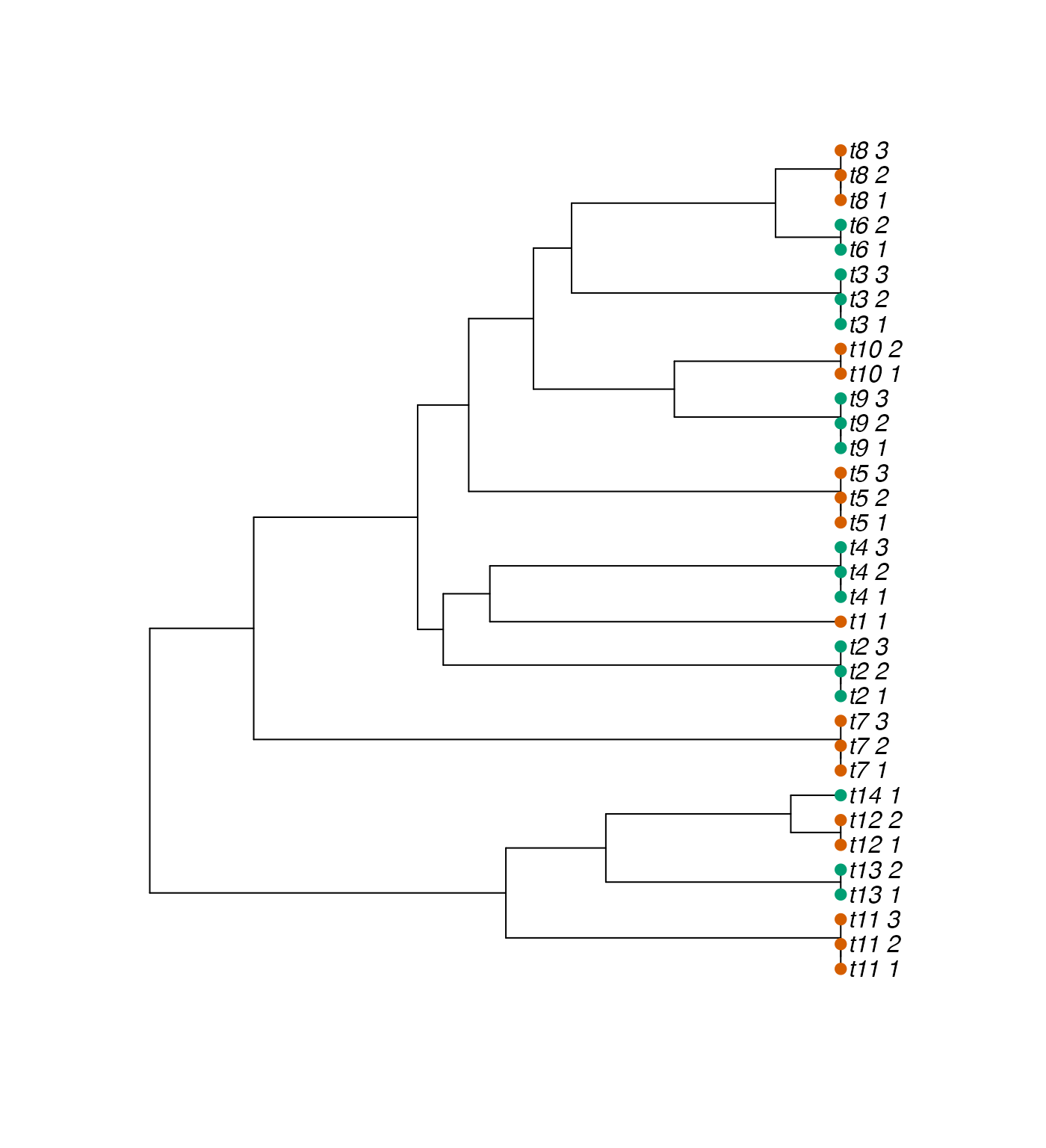
Phylogenetic tree with species and samples, with two conditions
Simulating data
Using this tree with associated condition design, we can then
generate a dataset using a “phylogenetic Poisson Log Normal” (pPLN)
distribution. We use here a Brownian Motion (BM) model of evolution for
the latent phylogenetic log normal continuous trait, and assume that the
phylogenetic model accounts for
of the latent trait variance (i.e. there is an added uniform
intra-species variance representing
of the total latent trait variation). Using the "auto"
setup, the counts are simulated so that they match empirical moments
found in Stern and Crandall (2018). OG
lengths are also drawn from a pPLN model, so that their moments match
those of the empirical dataset of Stern and
Crandall (2018). We choose to simulate
OGs,
of which are differentially expressed, with an effect size of
.
The following code creates a phyloCompData object
containing the simulated data set and saves it to a file named
"alt_BM_repl1.rds".
set.seed(12890926)
alt_BM <- generateSyntheticData(dataset = "alt_BM",
n.vars = 2000, samples.per.cond = 17,
n.diffexp = 200, repl.id = 1,
seqdepth = 1e7, effect.size = 3,
fraction.upregulated = 0.5,
output.file = "alt_BM_repl1.rds",
## Phylogenetic parameters
tree = tree, ## Phylogenetic tree
id.species = id_species, ## Species structure of samples
id.condition = id_cond, ## Condition design
model.process = "BM", ## The latent trait follows a BM
prop.var.tree = 0.9, ## Tree accounts for 90% of the variance
lengths.relmeans = "auto", ## OG length mean and dispersion
lengths.dispersions = "auto") ## are taken from an empirical exempleThe summarizeSyntheticDataSet works the same way as in
the base compcodeR package,
generating a report that summarize all the parameters used in the
simulation, and showing some diagnostic plots.
summarizeSyntheticDataSet(data.set = "alt_BM_repl1.rds",
output.filename = "alt_BM_repl1_datacheck.html")When applied to a phyloCompData object, it provides some
extra diagnostics, related to the phylogenetic nature of the data. In
particular, it contains MA-plots with TPM-normalized expression levels
to take OG length into account, which generally makes the original
signal clearer.
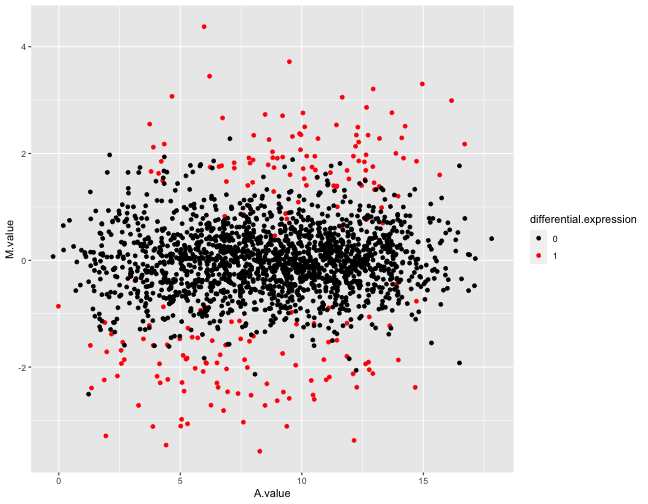
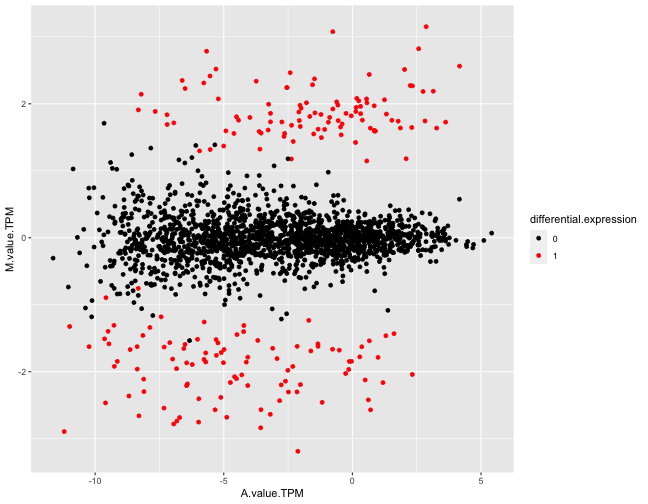
Example figures from the summarization report generated for a simulated data set. The top panel shows an MA plot, with the genes colored by the true differential expression status. The bottom panel shows the same plot, but using TPM-normalized estimated expression levels.
It also shows a log2 normalized counts heatmap plotted along the phylogeny, illustrating the phylogenetic structure of the differentially expressed OGs.
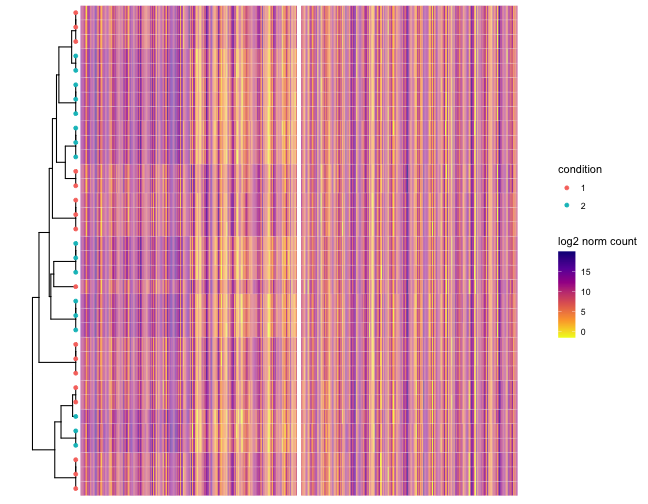
Example figures from the summarization report generated for a simulated data set. The tips colored by true differential expression status. Only the first 400 genes are represented. The first block of 200 genes are differencially expressed between condition 1 and 2. The second block of 200 genes are not differencially expressed.
Performing differential expression analysis
Differential expression analysis can be conducted using the same
framework used in the compcodeR
package, through the runDiffExp function.
All the standard methods can be used. To account for the phylogenetic nature of the data and for the varying length of the OGs, some methods have been added to the pool.
The code below applies three differential expression methods to the
data set generated above: the DESeq2
method adapted for varying lengths, the log2(TPM)
transformation for length normalization, combined with limma,
using the trend empirical Bayes correction, and accounting
for species-related correlations, and the phylogenetic regression tool
phylolm
applied on the same log2(TPM).
runDiffExp(data.file = "alt_BM_repl1.rds",
result.extent = "DESeq2", Rmdfunction = "DESeq2.createRmd",
output.directory = ".",
fit.type = "parametric", test = "Wald")
runDiffExp(data.file = "alt_BM_repl1.rds",
result.extent = "lengthNorm.limma", Rmdfunction = "lengthNorm.limma.createRmd",
output.directory = ".",
norm.method = "TMM",
length.normalization = "TPM",
data.transformation = "log2",
trend = FALSE, block.factor = "id.species")
runDiffExp(data.file = "alt_BM_repl1.rds",
result.extent = "phylolm", Rmdfunction = "phylolm.createRmd",
output.directory = ".",
norm.method = "TMM",
model = "BM", measurement_error = TRUE,
extra.design.covariates = NULL,
length.normalization = "TPM",
data.transformation = "log2")As for a regular compcodeR
analysis, example calls are provided in the reference manual (see the
help pages for the runDiffExp function), and a list of all
available methods can be obtained with the listcreateRmd()
function.
listcreateRmd()
#> [1] "DESeq2.createRmd"
#> [2] "DESeq2.length.createRmd"
#> [3] "DSS.createRmd"
#> [4] "EBSeq.createRmd"
#> [5] "edgeR.exact.createRmd"
#> [6] "edgeR.GLM.createRmd"
#> [7] "lengthNorm.limma.createRmd"
#> [8] "lengthNorm.sva.limma.createRmd"
#> [9] "logcpm.limma.createRmd"
#> [10] "NBPSeq.createRmd"
#> [11] "NOISeq.prenorm.createRmd"
#> [12] "phylolm.createRmd"
#> [13] "sqrtcpm.limma.createRmd"
#> [14] "TCC.createRmd"
#> [15] "ttest.createRmd"
#> [16] "voom.limma.createRmd"
#> [17] "voom.ttest.createRmd"Comparing results from several differential expression methods
Given that the phyloCompData object has the same
structure with respect to the slots added by the differential expression
analysis (see the result
object, the procedure to compare results from several differential
expression methods is exactly the same as for a compData
object, and can be found in the corresponding
section section of the compcodeR
vignette.
Using your own data
As for a
compData object, it is still possible to input
user-defined data to produce a phyloCompData object for
differential expression methods comparisons. One only needs to provide
the additional information needed, that is the phylogenetic tree, and
the length matrix. The constructor method will make sure that the tree
is consistent with the count and length matrices, with the same
dimensions and consistent species names.
## Phylogentic tree with replicates
tree <- read.tree(text = "(((A1:0,A2:0,A3:0):1,B1:1):1,((C1:0,C2:0):1.5,(D1:0,D2:0):1.5):0.5);")
## Sample annotations
sample.annotations <- data.frame(
condition = c(1, 1, 1, 1, 2, 2, 2, 2), # Condition of each sample
id.species = c("A", "A", "A", "B", "C", "C", "D", "D") # Species of each sample
)
## Count Matrix
count.matrix <- round(matrix(1000*runif(8000), 1000))
## Length Matrix
length.matrix <- round(matrix(1000*runif(8000), 1000))
## Names must match
colnames(count.matrix) <- colnames(length.matrix) <- rownames(sample.annotations) <- tree$tip.label
## Extra infos
info.parameters <- list(dataset = "mydata", uID = "123456")
## Creation of the object
cpd <- phyloCompData(count.matrix = count.matrix,
sample.annotations = sample.annotations,
info.parameters = info.parameters,
tree = tree,
length.matrix = length.matrix)
## Check
check_phyloCompData(cpd)
#> [1] TRUEProviding your own differential expression code
To use your own differential expression code, you can follow the base
compcodeR instructions in the compcodeR
vignette.
The extended data object
The phylocompData data object is an S4 object that
extends the
compData object, with the following added slots:
tree[class phylo] (mandatory) – the phylogenetic tree describing the relationships between samples.length.matrix[class matrix] (mandatory) – the OG length matrix, with rows representing genes and columns representing samples.-
When produced with
generateSyntheticData, thesample.annotationsdata frame has added column:-
id.species[class characterornumeric] – the species for each sample. Should match with thetip.labelof thetreeslot.
-
-
When produced with
generateSyntheticData, thevariable.annotationsdata frame has an added columns:-
lengths.relmeans[class numeric] – the true mean values used in the simulations of the OG lengths. -
lengths.dispersions[class numeric] – the true dispersion values used in the simulations of the OG lengths. -
M.value.TPM[class numeric] – the estimated log2-fold change between conditions 1 and 2 for each OG using TPM length normalization. -
A.value.TPM[class numeric] – the estimated average expression in conditions 1 and 2 for each OG using TPM length normalization. -
prop.var.tree[class numeric] – the proportion of the variance explained by the phylogeny for each gene.
-
The same way as the compData object, the
phyloCompData object needs to be saved to a file with
extension .rds.
The evaluation metrics
The evaluation metrics are unchanged, and described in the corresponding section section of the compcodeR vignette.
Session info
sessionInfo()
#> R version 4.6.0 Patched (2026-04-27 r89967)
#> Platform: aarch64-apple-darwin23
#> Running under: macOS Sequoia 15.7.4
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.6/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.6/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: UTC
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets
#> [6] methods base
#>
#> other attached packages:
#> [1] ape_5.8-1 compcodeR_1.49.0 sm_2.2-6.0
#> [4] BiocStyle_2.39.0
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.2.1 timeDate_4052.112
#> [3] dplyr_1.2.1 farver_2.1.2
#> [5] S7_0.2.2 bitops_1.0-9
#> [7] fastmap_1.2.0 promises_1.5.0
#> [9] digest_0.6.39 rpart_4.1.27
#> [11] mime_0.13 lifecycle_1.0.5
#> [13] cluster_2.1.8.2 statmod_1.5.1
#> [15] ROCR_1.0-12 magrittr_2.0.5
#> [17] compiler_4.6.0 rlang_1.2.0
#> [19] sass_0.4.10 tools_4.6.0
#> [21] yaml_2.3.12 knitr_1.51
#> [23] htmlwidgets_1.6.4 statip_0.2.3
#> [25] RColorBrewer_1.1-3 KernSmooth_2.23-26
#> [27] timeSeries_4052.112 fBasics_4052.98
#> [29] desc_1.4.3 grid_4.6.0
#> [31] caTools_1.18.3 stabledist_0.7-2
#> [33] xtable_1.8-8 edgeR_4.9.9
#> [35] ggplot2_4.0.3 scales_1.4.0
#> [37] gtools_3.9.5 MASS_7.3-65
#> [39] cli_3.6.6 rmarkdown_2.31
#> [41] ragg_1.5.2 generics_0.1.4
#> [43] otel_0.2.0 cachem_1.1.0
#> [45] stringr_1.6.0 parallel_4.6.0
#> [47] stable_1.1.7 BiocManager_1.30.27
#> [49] vctrs_0.7.3 jsonlite_2.0.0
#> [51] bookdown_0.46 rmutil_1.1.10
#> [53] clue_0.3-68 systemfonts_1.3.2
#> [55] locfit_1.5-9.12 limma_3.67.3
#> [57] jquerylib_0.1.4 spatial_7.3-18
#> [59] glue_1.8.1 pkgdown_2.2.0.9000
#> [61] stringi_1.8.7 gtable_0.3.6
#> [63] later_1.4.8 shinydashboard_0.7.3
#> [65] tibble_3.3.1 pillar_1.11.1
#> [67] htmltools_0.5.9 gplots_3.3.0
#> [69] R6_2.6.1 textshaping_1.0.5
#> [71] vioplot_0.5.1 evaluate_1.0.5
#> [73] shiny_1.13.0 lattice_0.22-9
#> [75] markdown_2.0 png_0.1-9
#> [77] modeest_2.4.0 httpuv_1.6.17
#> [79] bslib_0.10.0 Rcpp_1.1.1-1.1
#> [81] nlme_3.1-169 xfun_0.57
#> [83] fs_2.1.0 zoo_1.8-15
#> [85] pkgconfig_2.0.3