Comparing methods for differential expression analysis of RNAseq data with the compcodeR package
Charlotte Soneson
2026-04-28
Source:vignettes/compcodeR.Rmd
compcodeR.Rmd
library(compcodeR)
#> Loading required package: sm
#> Package 'sm', version 2.2-6.0: type help(sm) for summary informationIntroduction
compcodeR is an R package that provides an interface to several popular methods for differential expression analysis of RNAseq data and contains functionality for comparing the outcomes and performances of several differential expression methods applied to the same data set. The package also contains a function for generating synthetic RNAseq counts, using the simulation framework described in more detail in Soneson and Delorenzi (2013).
This vignette provides a tutorial on how to use the different functionalities of the compcodeR package.
Currently, the differential expression interfaces provided in the package are restricted to comparisons between two conditions. However, many of the comparison functions are more general and can also be applied to test results from other contrast types, as well as to test results from other data types than RNAseq.
Important! Since compcodeR
provides interfaces to differential expression analysis methods
implemented in other R packages, take care to cite the appropriate
references if you use any of the interface functions (see the reference
manual for more information). Also be sure to always check the code that
was used to run the differential expression analysis, using e.g. the
generateCodeHTMLs() function (see below for more
information) to make sure that parameters etc. agree with your
intentions and that there were no errors or serious warnings. These
interface functions are provided for convenience and as templates for
other, user-defined workflows, and there is no guarantee that the
included differential expression code is kept up-to-date with the latest
recommendations and best practices for running each of the interfaced
methods, or that the chosen settings are suitable in all situations, and
the user should make sure that the analysis is performed in the way they
intend. For instructions on how to add your own DE method, see below.
The compData class
Within the compcodeR
package (version >= 0.2.0), data sets and results are represented as
objects of the compData class. The functions in the package
are still compatible with the list-based representation used in version
0.1.0, but we strongly encourage users to use the compData
class, and all results generated by the package will be given in this
format. If you have a data or result object generated with compcodeR
version 0.1.0, you can convert it to a compData object
using the convertListTocompData function.
A compData object has at least three slots, containing
the count matrix, sample annotations and a list containing at least an
identifying name and a unique ID for the data set. It can also contain
variable annotations, such as information regarding genes that are known
to be differentially expressed. After performing a differential
expression analysis, the compData object contains
additional information, such as which method was used to perform the
analysis, which settings were used and the gene-wise results from the
analysis. More detailed information about the compData
class are available in the sections on the
data object and the result
object.
A sample workflow
This section contains a sample workflow showing the main functionalities of the compcodeR package. We start by generating a synthetic count data set, to which we then apply three different differential expression methods. Finally, we compare the outcome of the three methods and generate a report summarizing the results.
Simulating data
The simulations are performed following the description by Soneson and Delorenzi (2013). As an example, we
use the generateSyntheticData function to generate a
synthetic count data set containing 12,500 genes and two groups of 5
samples each, where 10% of the genes are simulated to be differentially
expressed between the two groups (equally distributed between up- and
downregulated in group 2 compared to group 1). Furthermore, the counts
for all genes are simulated from a Negative Binomial distribution with
the same dispersion in the two sample groups, and no outlier counts are
introduced. We filter the data set by excluding only the genes with zero
counts in all samples (i.e., those for which the total count is 0). This
simulation setting corresponds to the one denoted
in Soneson and Delorenzi (2013). The
following code creates a compData object containing the
simulated data set and saves it to a file named
"B_625_625_5spc_repl1.rds".
B_625_625 <- generateSyntheticData(dataset = "B_625_625", n.vars = 12500,
samples.per.cond = 5, n.diffexp = 1250,
repl.id = 1, seqdepth = 1e7,
fraction.upregulated = 0.5,
between.group.diffdisp = FALSE,
filter.threshold.total = 1,
filter.threshold.mediancpm = 0,
fraction.non.overdispersed = 0,
output.file = "B_625_625_5spc_repl1.rds")The summarizeSyntheticDataSet function provides
functionality to check some aspects of the simulated data by generating
a report summarizing the parameters that were used for the simulation,
as well as including some diagnostic plots. The report contains two
MA-plots, showing the estimated average expression level and the
log-fold change for all genes, indicating either the truly
differentially expressed genes or the total number of outliers
introduced for each gene. It also shows the log-fold changes estimated
from the simulated data versus those underlying the simulation. The
input to the summarizeSyntheticDataSet function can be
either a compData object or the path to a file containing
such an object.
summarizeSyntheticDataSet(data.set = "B_625_625_5spc_repl1.rds",
output.filename = "B_625_625_5spc_repl1_datacheck.html")Figure 1 shows two of the figures generated by this function. The top panel shows an MA plot with the genes colored by the true differential expression status. The bottom panel shows the relationship between the true log-fold changes between the two sample groups underlying the simulation, and the estimated log-fold changes based on the simulated counts.
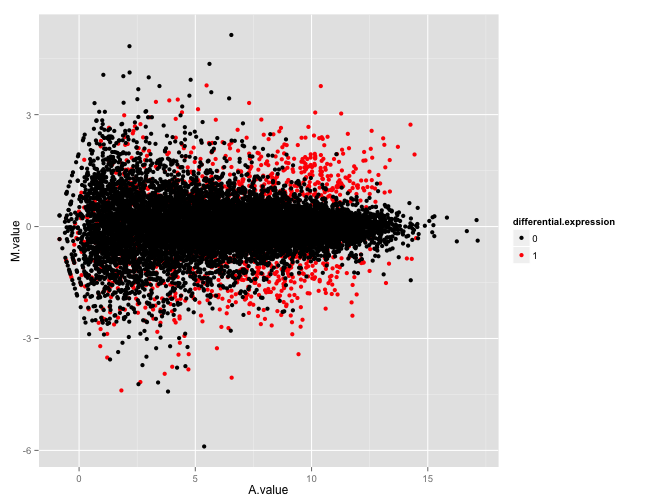
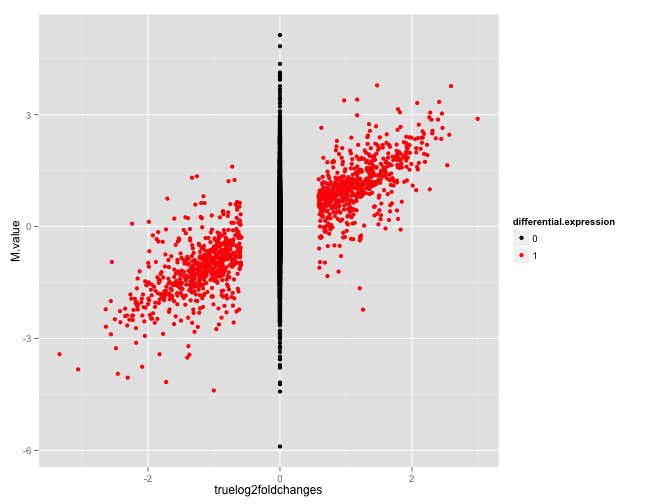
Example figures from the summarization report generated for a simulated data set. The top panel shows an MA plot, with the genes colored by the true differential expression status. The bottom panel shows the relationship between the true log-fold changes between the two sample groups underlying the simulation, and the estimated log-fold changes based on the simulated counts. Also here, the genes are colored by the true differential expression status.
Performing differential expression analysis
We will now apply some of the interfaced differential expression
methods to find genes that are differentially expressed between the two
conditions in the simulated data set. This is done through the
runDiffExp function, which is the main interface for
performing differential expression analyses in compcodeR.
The code below applies three differential expression methods to the data
set generated above: the voom transformation from the
limma
package (combined with limma for differential expression),
the exact test from the edgeR
package, and a regular t-test applied directly on the count level
data.
runDiffExp(data.file = "B_625_625_5spc_repl1.rds",
result.extent = "voom.limma", Rmdfunction = "voom.limma.createRmd",
output.directory = ".", norm.method = "TMM")
runDiffExp(data.file = "B_625_625_5spc_repl1.rds",
result.extent = "edgeR.exact", Rmdfunction = "edgeR.exact.createRmd",
output.directory = ".", norm.method = "TMM",
trend.method = "movingave", disp.type = "tagwise")
runDiffExp(data.file = "B_625_625_5spc_repl1.rds", result.extent = "ttest",
Rmdfunction = "ttest.createRmd",
output.directory = ".", norm.method = "TMM")The code needed to perform each of the analyses is provided in the
*.createRmd functions. To obtain a list of all available
*.createRmd functions (and hence of the available
differential expression methods), we can use the
listcreateRmd() function. Example calls are also provided
in the reference manual (see the help pages for the
runDiffExp function).
listcreateRmd()
#> [1] "DESeq2.createRmd"
#> [2] "DESeq2.length.createRmd"
#> [3] "DSS.createRmd"
#> [4] "EBSeq.createRmd"
#> [5] "edgeR.exact.createRmd"
#> [6] "edgeR.GLM.createRmd"
#> [7] "lengthNorm.limma.createRmd"
#> [8] "lengthNorm.sva.limma.createRmd"
#> [9] "logcpm.limma.createRmd"
#> [10] "NBPSeq.createRmd"
#> [11] "NOISeq.prenorm.createRmd"
#> [12] "phylolm.createRmd"
#> [13] "sqrtcpm.limma.createRmd"
#> [14] "TCC.createRmd"
#> [15] "ttest.createRmd"
#> [16] "voom.limma.createRmd"
#> [17] "voom.ttest.createRmd"You can also apply your own differential expression method to the simulated data.
Comparing results from several differential expression methods
Once we have obtained the results of the differential expression analyses (either by the methods interfaced by compcodeR or in other ways), we can compare the results and generate an HTML report summarizing the results from the different methods from many different aspects (see below for an overview of the comparison metrics). In compcodeR, there are two ways of invoking the comparison functionality; either directly from the command line or via a graphical user interface (GUI). The GUI is mainly included to avoid long function calls and provide a clear overview of the available methods and parameter choices. To use the GUI the R package rpanel must be installed (which assumes that BWidget is available). Moreover, the GUI may have rendering problems on certain platforms, particularly on small screens and if many methods are to be compared. Below, we will show how to perform the comparison using both approaches.
The graphical user interface
First, we consider the runComparisonGUI function, to
which we provide a list of directories containing our result files, and
the directory where the final report will be generated. Since the three
result files above were saved in the current working directory, we can
run the following code to perform the comparison:
runComparisonGUI(input.directories = ".",
output.directory = ".", recursive = FALSE)This opens a graphical user interface (Figure 2) where we can select which of the data files available in the input directories that should be included as a basis for the comparison, and which comparisons to perform. Through this interface, we can also set p-value cutoffs for significance and other parameters that will govern the behaviour of the comparison.
Important! When you have modified a value in one of the textboxes (p-value cutoffs etc.), press Return on your keyboard to confirm the assignment of the new value to the parameter. Always check in the resulting comparison report that the correct values were recognized and used for the comparisons.
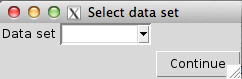
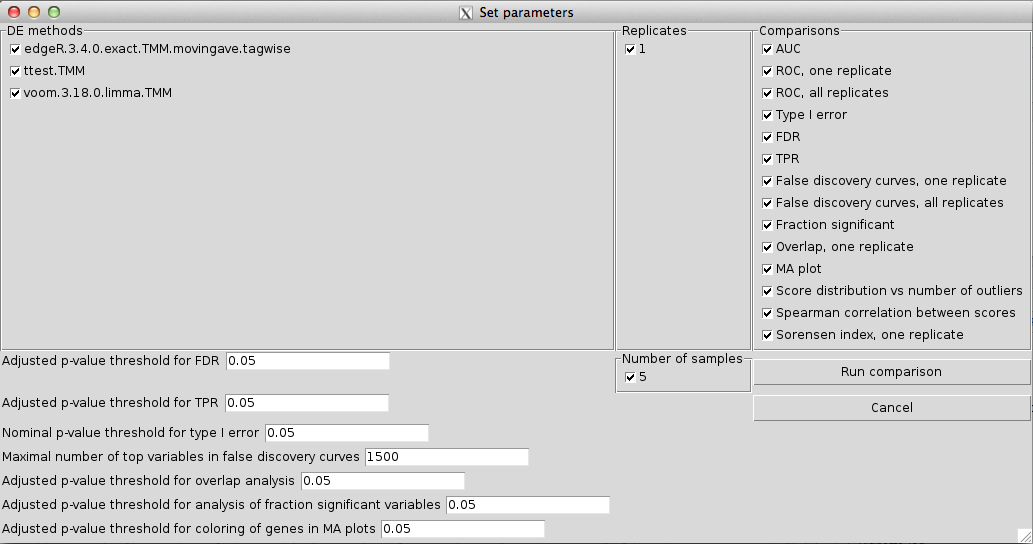
Screenshot of the graphical user interface used to select data set (left) and set parameters (right) for the comparison of differential expression methods. The available choices for the Data set, DE methods, Number of samples and Replicates are automatically generated from the compData objects available in the designated input directories. Only one data set can be used for the comparison. In the lower part of the window we can set (adjusted) p-value thresholds for each comparison method separately. For example, we can evaluate the true FDR at one adjusted p-value threshold, and estimate the TPR for another adjusted p-value threshold. We can also set the maximal number of top-ranked variables that will be considered for the false discovery curves.
After the selections have been made, the function will perform the
comparisons and generate an HTML report, which is saved in the
designated output directory and automatically named
compcodeR_report_<timestamp>.html. Please note that
depending on the number of compared methods and the number of included
data sets, this step may take several minutes. compcodeR
will notify you when the report is ready (with a message
Done! in the console).
Note! Depending on the platform you use to run R, you may see a prompt ($">"$) in the console before the analysis is done. However, compcodeR will always notify you when it is finished, by typing Done! in the console.
The comparison function will also generate a subdirectory called
compcodeR_code, where the R code used to perform each of
the comparisons is detailed in HTML reports, and another subdirectory
called compcodeR_figure, where the plots generated in the
comparison are saved. The code HTML reports can also be generated
manually for a given result file (given that the code
component is present in the compData object), using the
function generateCodeHTMLs. For example, to generate a
report containing the code that was run to perform the t-test above, as
well as the output from the R console, we can write:
generateCodeHTMLs("B_625_625_5spc_repl1_ttest.rds", ".")These reports are useful to check that there were no errors or warnings when running the differential expression analyses.
Direct command-line call
Next, we show how to run the comparison by calling the function
runComparison directly. In this case, we need to supply the
function with a list of result files to use as a basis for the
comparison. We can also provide a list of parameters (p-value
thresholds, the differential expression methods to include in the
comparison, etc.). The default values of these parameters are outlined
in the reference manual. The following code provides an example, given
the data generated above.
file.table <- data.frame(input.files = c("B_625_625_5spc_repl1_voom.limma.rds",
"B_625_625_5spc_repl1_ttest.rds",
"B_625_625_5spc_repl1_edgeR.exact.rds"),
stringsAsFactors = FALSE)
parameters <- list(incl.nbr.samples = NULL, incl.replicates = NULL,
incl.dataset = "B_625_625", incl.de.methods = NULL,
fdr.threshold = 0.05, tpr.threshold = 0.05,
typeI.threshold = 0.05, ma.threshold = 0.05,
fdc.maxvar = 1500, overlap.threshold = 0.05,
fracsign.threshold = 0.05,
comparisons = c("auc", "fdr", "tpr", "ma", "correlation"))
runComparison(file.table = file.table, parameters = parameters, output.directory = ".")By setting incl.nbr.samples,
incl.replicates and incl.de.methods to
NULL, we ask compcodeR
to include all results provided in the file.table. By
providing a vector of values for each of these variables, it is possible
to limit the selection to a subset of the provided files. Please note
that the values given to incl.replicates and
incl.nbr.samples are matched with values of the
info.parameters$repl.id and
info.parameters$samples.per.cond slots in the data/result
objects, and that the values given to incl.de.methods are
matched with values stored in the method.names$full.name
slot of the result objects. If the values do not match, the
corresponding result object will not be considered in the comparison.
Consult the package manual for the full list of comparison methods
available for use with the runComparison function. Setting
parameters = NULL implies that all results provided in the
file.table are used, and that all parameter values are set
to their defaults (see the reference manual). Note that only one dataset
identifier can be provided to the comparison (that is,
parameters$incl.dataset must be a single string).
Using your own data
The compcodeR
package provides a straightforward function for simulating count data
(generateSyntheticData). However, it is easy to apply the
interfaced differential expression methods to your own data, given that
it is provided in a compData object (see below for a description of the data format).
You can use the function check_compData to check that your
object satisfies the necessary criteria to be fed into the differential
expression methods.
To create a compData object from a count matrix and a
data frame with sample annotations, you can use the function
compData. The following code provides a minimal example.
Note that you need to provide a dataset name (a
‘description’ of the simulation settings) as well as a unique data set
identifier uID, which has to be unique for each
compData object (e.g., for each simulation instance, even
if the same simulation parameters are used).
count.matrix <- matrix(round(1000*runif(4000)), 1000, 4)
sample.annot <- data.frame(condition = c(1, 1, 2, 2))
info.parameters <- list(dataset = "mytestdata", uID = "123456")
cpd <- compData(count.matrix = count.matrix,
sample.annotations = sample.annot,
info.parameters = info.parameters)
check_compData(cpd)
#> [1] TRUEProviding your own differential expression code
The compcodeR package provides an interface for calling some of the most commonly used differential expression methods developed for RNAseq data. However, it is easy to incorporate your own favorite method. In principle, this can be done in one of two ways:
-
Write a
XXX.createRmdfunction (whereXXXcorresponds to your method), similar to the ones provided in the package, which creates a.Rmdfile containing the code that is run to perform the differential expression analysis. Then call this function through therunDiffExpfunction. When implementing your function, make sure that the output is acompDataobject, structured as described below. The*.createRmdfunctions provided in the package take the following input arguments:-
data.path– the path to the.rdsfile containing thecompDataobject to which the differential expression will be applied. -
result.path– the path to the.rdsfile where the resultingcompDataobject will be stored. -
codefile– the name of the code (with extension .Rmd) where the code will be stored. - any arguments for setting parameters of the differential expression analysis.
-
Run the differential expression analysis completely outside the compcodeR package, and save the result in a
compDataobject with the slots described below.
You can use the check_compData_results function to check
if your object satisfies the necessary conditions for being used as the
output of a differential expression analysis and compared to results
obtained by other methods with compcodeR.
The format of the data and result objects
This section details the format of the data and result objects
generated and used by the compcodeR
package. Both objects are of the compData class. The format
guidelines below must be be followed if you apply the functions in the
package to a data set of your own, or to differential expression results
generated outside the package. Note that for most of the functionality
of the package, the objects should be saved separately to files with a
.rds extension, and the path of this object is provided to
the functions.
The data object
The compData data object used by compcodeR
is an S4 object with the following slots:
count.matrix[class matrix] (mandatory) – the count matrix, with rows representing genes and columns representing samples.-
sample.annotations[class data.frame] (mandatory) – sample annotations. Each row corresponds to one sample, and each column to one annotation. The data objects generated by thegenerateSyntheticDatafunction have two annotations:-
condition[class characterornumeric] (mandatory) – the class for each sample. Currently the differential expression implementations in the package supports only two-group comparisons, hence theconditionshould have exactly two unique values. -
depth.factor[class numeric] – the depth factor for each sample. This factor, multiplied by thesequencing depth, corresponds to the target library size of the sample when simulating the counts.
-
-
info.parameters[class list] – a list detailing the parameter values that have been used for the simulation.-
dataset[class character] (mandatory) – the name of the data set. -
samples.per.cond[class numeric] – the number of samples in each of the two conditions. -
n.diffexp[class numeric] – the number of genes that are simulated to be differentially expressed between the two conditions. -
repl.id[class numeric] – a replicate number, which can be set to differentiate between different instances generated with the exact same simulation settings. -
seqdepth[class numeric] – the “base” sequencing depth that was used for the simulations. For each sample, it is modified by multiplication with a value sampled uniformly betweenminfactandmaxfactto generate the actual sequencing depth for the sample. -
minfact[class numeric] – the lower bound on the values used to multiply theseqdepthto generate the actual sequencing depth for the individual samples. -
maxfact[class numeric] – the upper bound on the values used to multiply theseqdepthto generate the actual sequencing depth for the individual samples. -
fraction.upregulated[class numeric] – the fraction of the differentially expressed genes that were simulated to be upregulated in condition 2 compared to condition 1. Must be in the interval [0, 1]. The remaining differentially expressed genes are simulated to be downregulated in condition 2. -
between.group.diffdisp[class logical] – whether the counts from the two conditions were simulated with different dispersion parameters. -
filter.threshold.total[class numeric] – the filter threshold that is applied to the total count across all samples. All genes for which the total count is below this number have been excluded from the data set in the simulation process. -
filter.threshold.mediancpm[class numeric] – the filter threshold that is applied to the median count per million (cpm) across all samples. All genes for which the median cpm is below this number have been excluded from the data set in the simulation process. -
fraction.non.overdispersed[class numeric] – the fraction of the genes in the data set that are simulated according to a model without overdispersion (i.e., a Poisson model). -
random.outlier.high.prob[class numeric] – the fraction of extremely high random outlier counts introduced in the simulated data sets. Please consult Soneson and Delorenzi (2013) for a detailed description of the different types of outliers. -
random.outlier.low.prob[class numeric] – the fraction of extremely low random outlier counts introduced in the simulated data sets. Please consult Soneson and Delorenzi (2013) for a detailed description of the different types of outliers. -
single.outlier.high.prob[class numeric] – the fraction of extremely high single outlier counts introduced in the simulated data sets. Please consult Soneson and Delorenzi (2013) for a detailed description of the different types of outliers. -
single.outlier.low.prob[class numeric] – the fraction of extremely low single outlier counts introduced in the simulated data sets. Please consult Soneson and Delorenzi (2013) for a detailed description of the different types of outliers. -
effect.size[class numeric] – the degree of differential expression, i.e., a measure of the minimal effect size. Alternatively, a vector of provided effect sizes for each of the variables. -
uID[class character] (mandatory) – a unique identification number given to each data set. For the data sets simulated with compcodeR, it consists of a sequence of 10 randomly generated alpha-numeric characters.
-
filtering[class character] – a summary of the filtering that has been applied to the data.-
variable.annotations[class data.frame] – annotations for each of the variables in the data set. Each row corresponds to one variable, and each column to one annotation. No annotation is mandatory, however some of them are necessary if the differential expression results for the data are going to be used for certain comparison tasks. The data sets simulated within compcodeR have the following named variable annotations:-
truedispersions.S1[class numeric] – the true dispersions used in the simulations of the counts for the samples in condition 1. -
truedispersions.S2[class numeric] – the true dispersions used in the simulations of the counts for the samples in condition 2. -
truemeans.S1[class numeric] – the true mean values used in the simulations of the counts for the samples in condition 1. -
truemeans.S2[class numeric] – the true mean values used in the simulations of the counts for the samples in condition 2. -
n.random.outliers.up.S1[class numeric] – the number of extremely high random outliers introduced for each gene in condition 1. -
n.random.outliers.up.S2[class numeric] – the number of extremely high random outliers introduced for each gene in condition 2. -
n.random.outliers.down.S1[class numeric] – the number of extremely low random outliers introduced for each gene in condition 1. -
n.random.outliers.down.S2[class numeric] – the number of extremely low random outliers introduced for each gene in condition 2. -
n.single.outliers.up.S1[class numeric] – the number of extremely high single outliers introduced for each gene in condition 1. -
n.single.outliers.up.S2[class numeric] – the number of extremely high single outliers introduced for each gene in condition 2. -
n.single.outliers.down.S1[class numeric] – the number of extremely low single outliers introduced for each gene in condition 1. -
n.single.outliers.down.S2[class numeric] – the number of extremely low single outliers introduced for each gene in condition 2. -
M.value[class numeric] – the estimated log2-fold change between conditions 1 and 2 for each gene. These values were estimated using the edgeR package. -
A.value[class numeric] – the estimated average expression in conditions 1 and 2 for each gene. These values were estimated using the edgeR package. -
truelog2foldchanges[class numeric] – the “true” log2-fold changes between conditions 1 and 2 for each gene, based on the parameters used for simulation. -
upregulation[class numeric] – a binary annotation (0/1) indicating which genes are simulated to be upregulated in condition 2 compared to condition 1. (Upregulated genes are indicated with a 1.) -
downregulation[class numeric] – a binary annotation (0/1) indicating which genes are simulated to be downregulated in condition 2 compared to condition 1. (Downregulated genes are indicated with a 1.) -
differential.expression[class numeric] (mandatory for many comparisons, such as the computation of false discovery rates, true positive rates, ROC curves, false discovery curves etc.)– a binary annotation (0/1) indicating which genes are simulated to be differentially expressed in condition 2 compared to condition 1. In other words, the sum ofupregulationanddownregulation.
-
To apply the functions of the package to a compData
object of the type detailed above, it needs to be saved to a file with
extension .rds. To save the object cpd to the
file saveddata.rds, simply type
saveRDS(cpd, "saveddata.rds")The result object
When applying one of the differential expression methods interfaced
through compcodeR,
the compData object is extended with some additional slots.
These are described below.
analysis.date[class character] – the date and time when the differential expression analysis was performed.package.version[class character] – the version of the packages used in the differential expression analysis.-
method.names[class list] (mandatory) – a list containing the names of the differential expression method, which should be used to identify the results in the comparison. Contains two components:-
short.name[class character] – a short name, used for convenience. -
full.name[class character] – a fully identifying name of the differential expression method, including e.g. version numbers and parameter values. In the method comparisons, the results will be grouped based on thefull.name.
-
code[class character] – the code that was used to run the differential expression analysis, in R markdown (.Rmd) format.-
result.table[class data.frame] (mandatory) – a data frame containing the results of the differential expression analysis. Each row corresponds to one gene, and each column to a value generated by the analysis. The precise columns will depend on the method applied. The following columns are used by at least one of the methods interfaced by compcodeR:-
pvalue[class numeric] – the nominal p-values. -
adjpvalue[class numeric] – p-values adjusted for multiple comparisons. -
logFC[class numeric] – estimated log-fold changes between the two conditions. -
score[class numeric] (mandatory) – the score that will be used to rank the genes in order of significance. Note that high scores always signify differential expression, that is, a strong association with the predictor. For example, for methods returning a nominal p-value the score is generally obtained as1 - pvalue. -
FDR[class numeric] – the false discovery rate estimate. -
posterior.DE[class numeric] – the posterior probability of differential expression. -
prob.DE[class numeric] – The conditional probability of differential expression. -
lfdr[class numeric] – the local false discovery rate. -
statistic[class numeric] – a test statistic from the differential expression analysis. -
dispersion.S1[class numeric] – dispersion estimate in condition 1. -
dispersion.S2[class numeric] – dispersion estimate in condition 2.
-
For many of the comparison methods, the naming of the result columns
is important. For example, the p-value column must be named
pvalue in order to be recognized by the comparison method
computing type I error. Similarly, either an adjpvalue or
an FDR column must be present in order to apply the
comparison methods requiring adjusted p-value/FDR cutoffs. If both are
present, the adjpvalue column takes precedence over the
FDR column.
To be used in the comparison function, the result
compData object must be saved to a .rds
file.
The evaluation metrics
This section provides an overview of the methods that are implemented
in compcodeR
for comparing differential expression results obtained by different
methods. The selection of which methods to apply is made through a
graphical user interface that is opened when the
runComparisonGUI function is called (see Figure 2).
Alternatively, the selection of methods can be supplied to the
runComparison function directly, to circumvent the GUI.
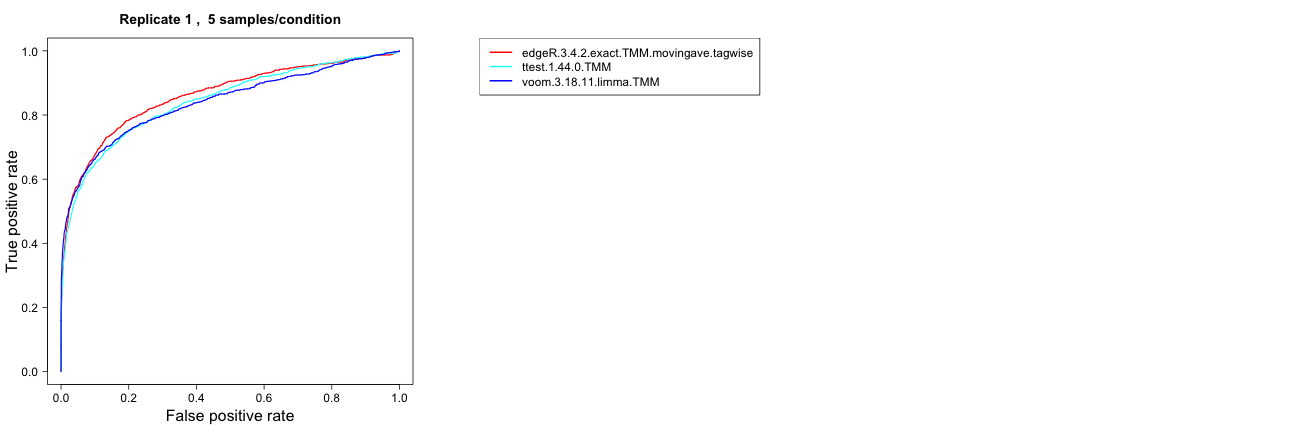
ROC (one replicate/all replicates)
This method computes a ROC curve for either a single representative
among the replicates of a given data set, or for all replicates (in
separate plots). The ROC curves are generated by plotting the true
positive rate (TPR, on the y-axis) versus the false positive rate (FPR,
on the x-axis) when varying the cutoff on the score (see
the description of the result object above). A good ranking method gives
a ROC curve which passes close to the upper left corner of the plot,
while a bad method gives a ROC curve closer to the diagonal. Calculation
of the ROC curves requires that the differential expression status of
each gene is provided.
AUC
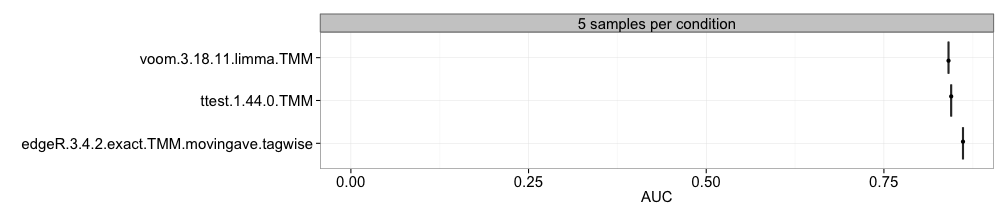
This comparison method computes the area under the ROC curve and represents the result in boxplots, where each box summarizes the results for one method across all replicates of a data set. A good ranking method gives to a large value of the AUC. This requires that the differential expression status of each gene is provided.
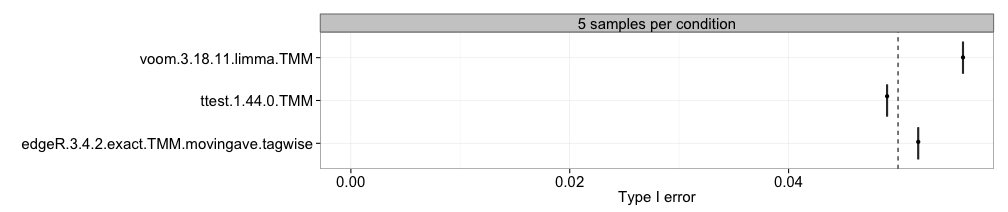
Type I error
This approach computes the type I error (the fraction of the genes
that are truly non-differentially expressed that are called significant
at a given nominal p-value threshold) for each method and sample size
separately, and represent it in boxplots, where each box summarizes the
results for one method across all replicates of a data set. This
requires that the differential expression status of each gene is
provided (see the description of the data object above) and that the
pvalue column is present in the result.table
of the included result objects. The dashed vertical line represents the
imposed nominal p-value threshold, and we wish that the observed type I
error is lower than this threshold.
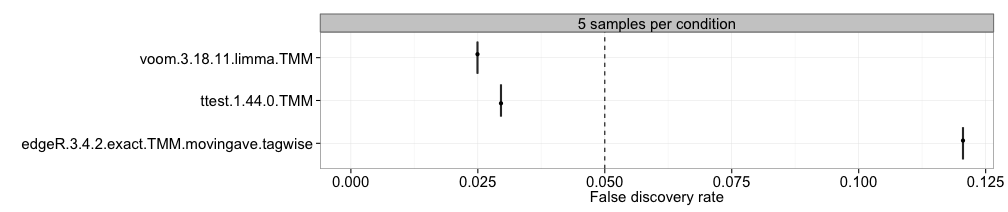
FDR
Here, we compute the observed false discovery rate (the fraction of
the genes called significant that are truly non-differentially expressed
at a given adjusted p-value/FDR threshold) for each method and sample
size separately, and represent the estimates in boxplots, where each box
summarizes the results for one method across all replicates of a data
set. This requires that the differential expression status of each gene
is provided (see the description of the data object above) and that the
adjpvalue or FDR column is present in the
result.table of the included result objects. The dashed
vertical line represents the imposed adjusted p-value threshold (that
is, the level at which we wish to control the false discovery rate).
FDR as a function of expression level
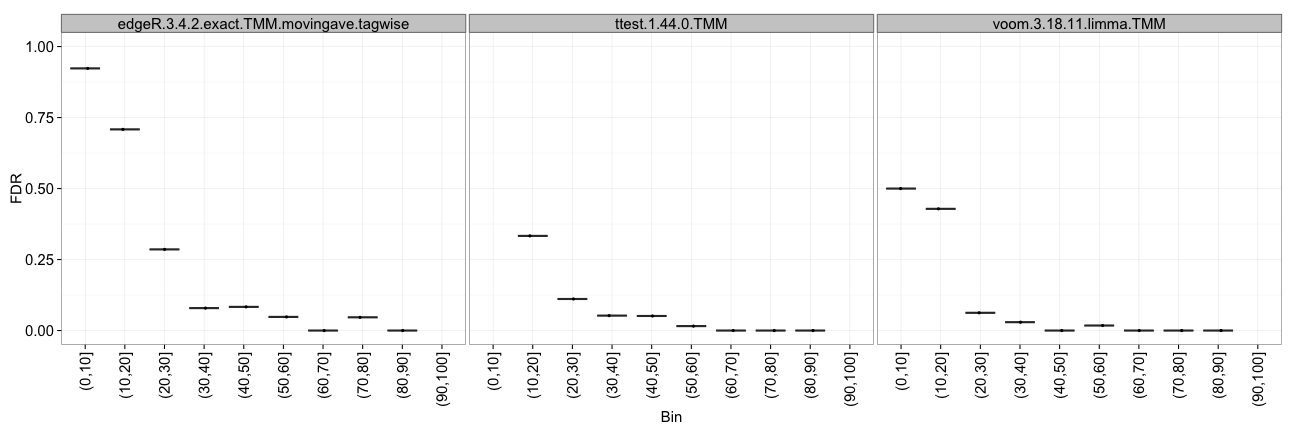
Instead of looking at the overall false discovery rate, this method allows us to study the FDR as a function of the average expression level of the genes. For each data set, the average expression levels are binned into 10 bins of equal size (i.e., each containing 10% of the genes), and the FDR is computed for each of them. The results are shown by means of boxplots, summarizing the results across all replicates of a data set.
TPR
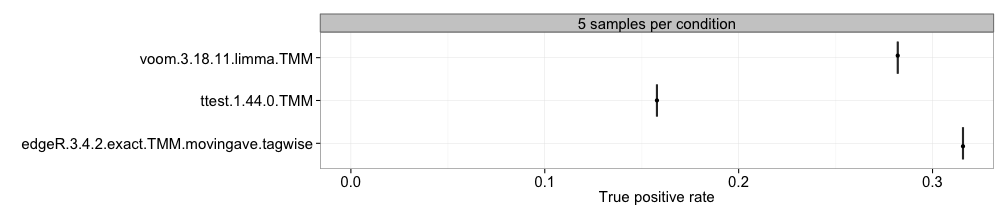
With this approach, we compute the observed true positive rate (the
fraction of the truly differentially expressed genes that are called
significant at a given adjusted p-value/FDR threshold) for each method
and sample size separately, and represent the estimates in boxplots,
where each box summarizes the results for one method across all
replicates of a data set. This requires that the differential expression
status of each gene is provided (see the description of the data object
above) and that the adjpvalue or FDR column is
present in the result.table of the included result objects.
There is often a trade-off between achieving a high TPR (which is
desirable) and controlling the number of false positives, and hence the
TPR plots should typically be interpreted together with the type I error
and/or FDR plots.
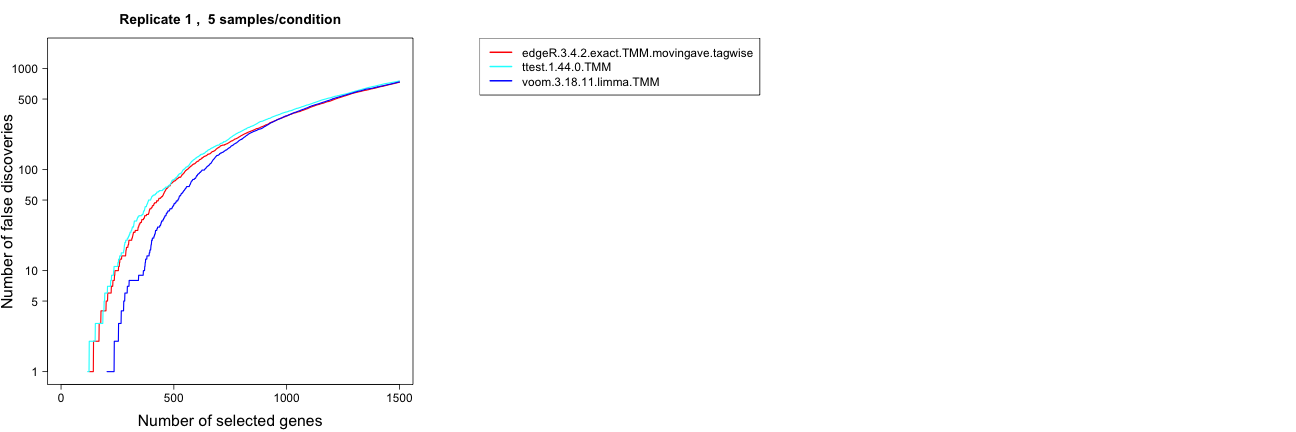
False discovery curves (one replicate/all replicates)
This choice plots the false discovery curves, depicting the number of
false discoveries (i.e., truly non-differentially expressed genes) that
are encountered when traversing the ranked list of genes ordered in
decreasing order by the score column of the
result.table. As for the ROC curves, the plots can be made
for a single replicate or for all replicates (each in a separate
figure). A well-performing method is represented by a slowly rising
false discovery curve (in other words, few false positives among the
top-ranked genes).
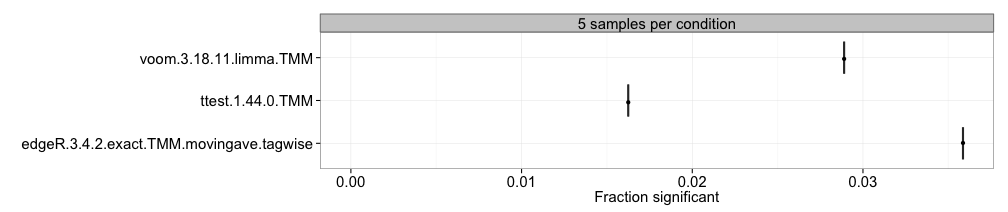
Fraction/number of significant genes
We can also compute the fraction and/or number of the genes that are
called significant at a given adjusted p-value/FDR threshold for each
method and sample size separately, and represent the estimates in
boxplots, where each box summarizes the results for one method across
all replicates of a data set. This requires that the
adjpvalue or FDR column is present in the
result.table of the included result objects. Since this
plot does not incorporate information about the number of the identity
of the truly differentially expressed genes, we can not conclude whether
a large or small fraction of significant genes is preferable. Thus, the
plot is merely an indication of which methods are more liberal (giving
more significantly differentially expressed genes) and which are more
conservative.
Overlap, one replicate
For each pair of methods, this approach computes the overlap between
the sets of genes called differentially expressed by each of them at a
given adjusted p-value/FDR threshold. Only one representative of the
replicates of a data set is used. This requires that the
adjpvalue or FDR column is present in the
result.table of the included result objects. Since the
overlap depends on the number of genes called differentially expressed
by the different methods, a normalized overlap measure (the Sorensen
index) is also provided (see below). The results are represented as a
table of the sizes of the overlaps between each pair of methods.
Sorensen index, one replicate
For each pair of methods, this approach computes a normalized
pairwise overlap value between the sets of genes called differentially
expressed by each of them at a given adjusted p-value/FDR threshold.
Only one representative of the replicates of a data set is used. This
requires that the adjpvalue or FDR column is
present in the result.table of the included result objects.
The Sorensen index (also called the Dice coefficient) for two sets
and
is given by
where
denotes the cardinality of a set (the number of elements in the set).
The results are represented in the form of a table, and also as a
colored heatmap, where the color represents the degree of overlap
between the sets of differentially expressed genes found by different
methods.
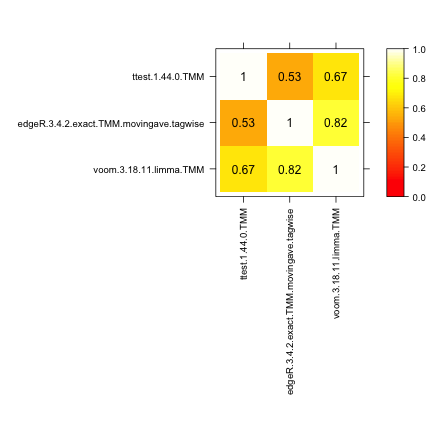
MA plot
With this approach we can construct MA-plots (one for each differential expression method) for one replicate of a data set, depicting the estimated log-fold change (on the y-axis) versus the estimated average expression (on the x-axis). The genes that are called differentially expressed are marked with color.
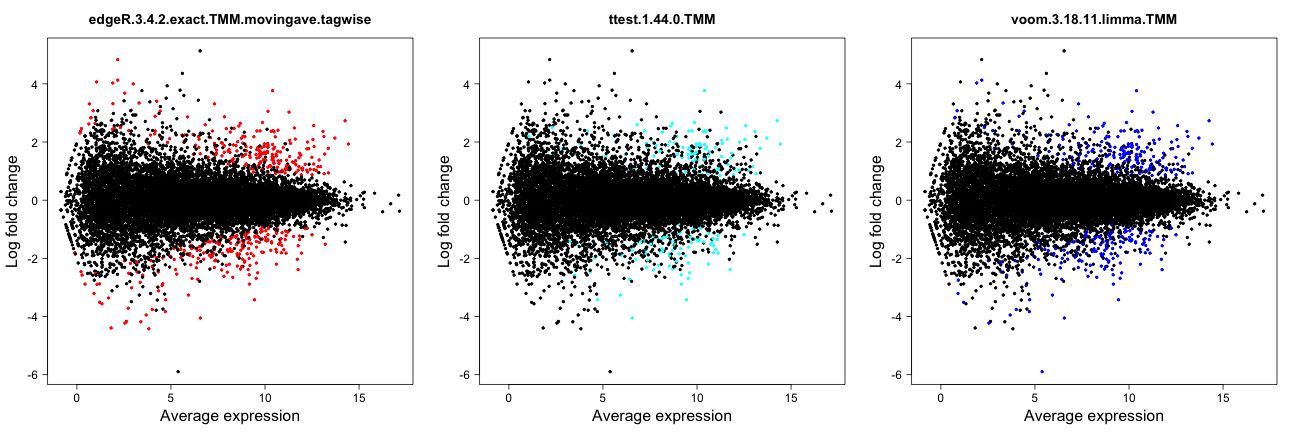
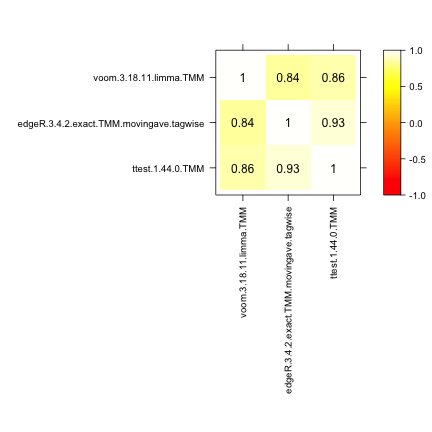
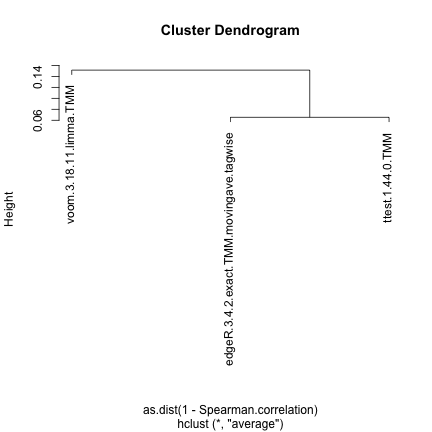
Spearman correlation
This approach computes the pairwise Spearman correlation between the
gene scores obtained by different differential expression methods (the
score component of the result.table). The
correlations are visualized by means of a color-coded table and used to
construct a dissimilarity measure for hierarchical clustering of the
differential expression methods.
Score distribution vs number of outliers
This method plots the distribution of the gene scores obtained by
different differential expression methods (the score
component of the result.table, see the description of the
result object above) as a function of the number of outliers imposed for
the genes. The distributions are visualized by means of violin plots. In
the example below, there are no outliers introduced. Recall that a high
score corresponds to more significant genes. In the presence of
outliers, the score distribution plots can be used to examine whether
the presence of an outlier count for a gene shifts the score
distribution towards higher or lower values.
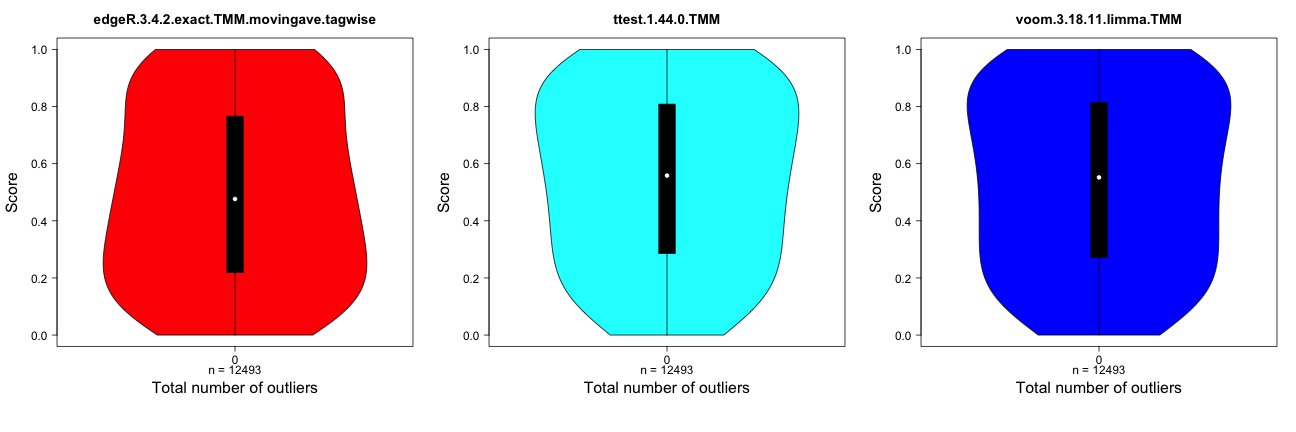
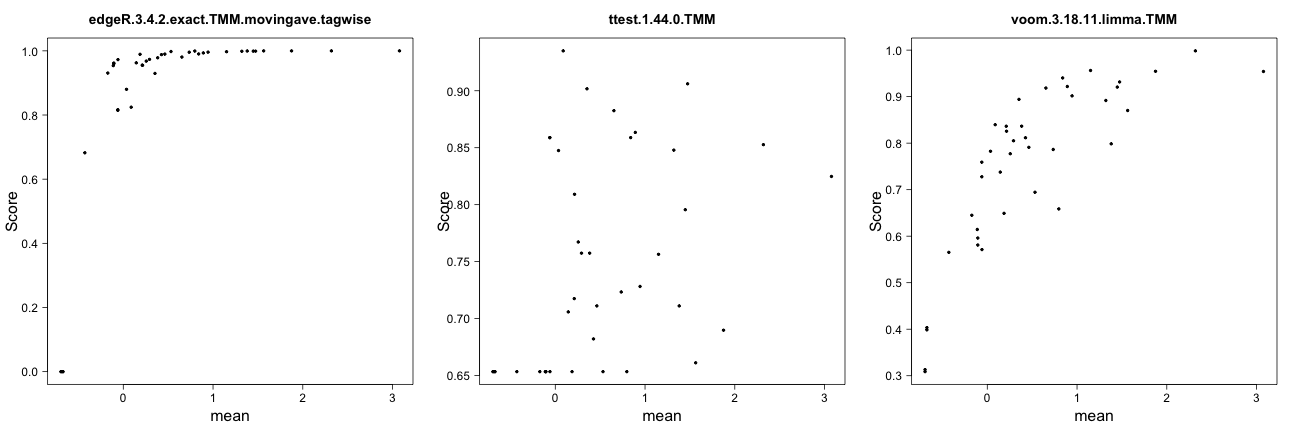
Score distribution vs average expression level
This method plots the gene scores (the score component
of the result.table, see the description of the result
object above) as a function of the average expression level of the gene.
Recall that high scores correspond to more significant genes. The
colored line shows the trend in the relationship between the two
variables by means of a loess fit. These plots can be used to examine
whether highly expressed genes tend to have (e.g.) higher scores than
lowly expressed genes, and more generally to provide a useful
characteristic of the methods. However, it is not clear what would be
the “optimal” behavior.

Score vs ‘signal’ for genes expressed in only one condition
This method plots the gene scores (the score component
of the result.table, see the description of the result
object above) as a function of the ‘signal strength’ for genes that are
detected in only one of the two conditions. The signal strength can be
defined either as the average (log-transformed) normalized pseudo-count
in the condition where the gene is expressed, or as the signal-to-noise
ratio in this condition, that is, as the average log-transformed
normalized pseudo-count divided by the standard deviation of the
log-transformed normalized pseudo-count. Typically, we would expect that
the score increases with the signal strength.
Matthew’s correlation coefficient
Matthew’s correlation coefficient summarizes the result of a classification task in a single number, incorporating the number of false positives, true positives, false negatives and true negatives. A correlation coefficient of +1 indicates perfect classification, while a correlation coefficient of -1 indicates perfect “anti-classification”, i.e., that all objects are misclassified.
Session info
sessionInfo()
#> R version 4.6.0 Patched (2026-04-27 r89967)
#> Platform: aarch64-apple-darwin23
#> Running under: macOS Sequoia 15.7.4
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.6/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.6/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: UTC
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets
#> [6] methods base
#>
#> other attached packages:
#> [1] compcodeR_1.49.0 sm_2.2-6.0 BiocStyle_2.39.0
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 xfun_0.57
#> [3] bslib_0.10.0 ggplot2_4.0.3
#> [5] htmlwidgets_1.6.4 caTools_1.18.3
#> [7] lattice_0.22-9 vctrs_0.7.3
#> [9] tools_4.6.0 bitops_1.0-9
#> [11] generics_0.1.4 tibble_3.3.1
#> [13] cluster_2.1.8.2 pkgconfig_2.0.3
#> [15] KernSmooth_2.23-26 RColorBrewer_1.1-3
#> [17] S7_0.2.2 desc_1.4.3
#> [19] lifecycle_1.0.5 stringr_1.6.0
#> [21] compiler_4.6.0 farver_2.1.2
#> [23] textshaping_1.0.5 gplots_3.3.0
#> [25] statmod_1.5.1 clue_0.3-68
#> [27] httpuv_1.6.17 htmltools_0.5.9
#> [29] sass_0.4.10 rmutil_1.1.10
#> [31] yaml_2.3.12 later_1.4.8
#> [33] pillar_1.11.1 pkgdown_2.2.0.9000
#> [35] jquerylib_0.1.4 MASS_7.3-65
#> [37] cachem_1.1.0 limma_3.67.3
#> [39] vioplot_0.5.1 rpart_4.1.27
#> [41] mime_0.13 fBasics_4052.98
#> [43] gtools_3.9.5 tidyselect_1.2.1
#> [45] locfit_1.5-9.12 digest_0.6.39
#> [47] stringi_1.8.7 dplyr_1.2.1
#> [49] bookdown_0.46 fastmap_1.2.0
#> [51] grid_4.6.0 cli_3.6.6
#> [53] magrittr_2.0.5 stable_1.1.7
#> [55] edgeR_4.9.9 ROCR_1.0-12
#> [57] promises_1.5.0 scales_1.4.0
#> [59] rmarkdown_2.31 otel_0.2.0
#> [61] timeDate_4052.112 png_0.1-9
#> [63] zoo_1.8-15 ragg_1.5.2
#> [65] timeSeries_4052.112 statip_0.2.3
#> [67] shiny_1.13.0 evaluate_1.0.5
#> [69] knitr_1.51 stabledist_0.7-2
#> [71] modeest_2.4.0 markdown_2.0
#> [73] rlang_1.2.0 Rcpp_1.1.1-1.1
#> [75] spatial_7.3-18 xtable_1.8-8
#> [77] glue_1.8.1 BiocManager_1.30.27
#> [79] shinydashboard_0.7.3 jsonlite_2.0.0
#> [81] R6_2.6.1 systemfonts_1.3.2
#> [83] fs_2.1.0